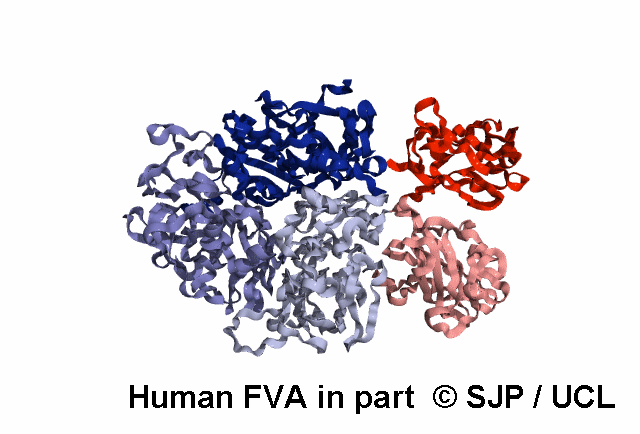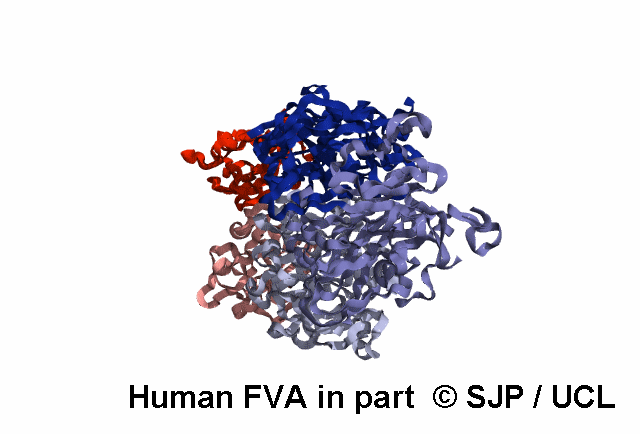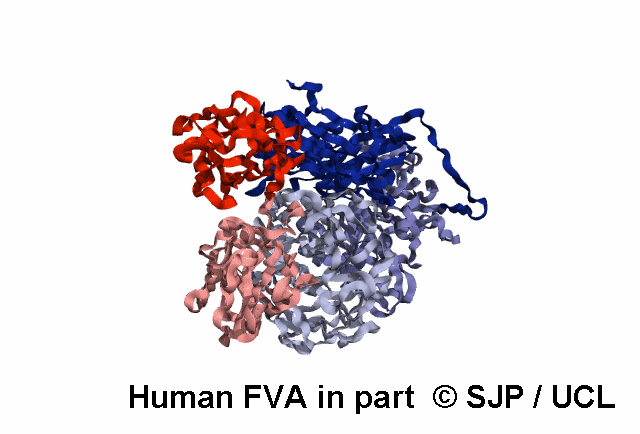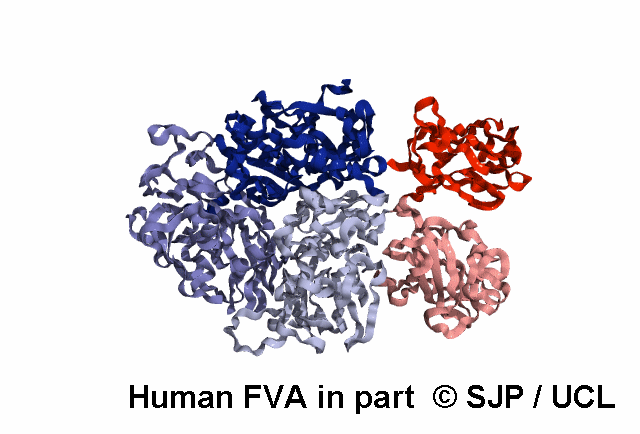
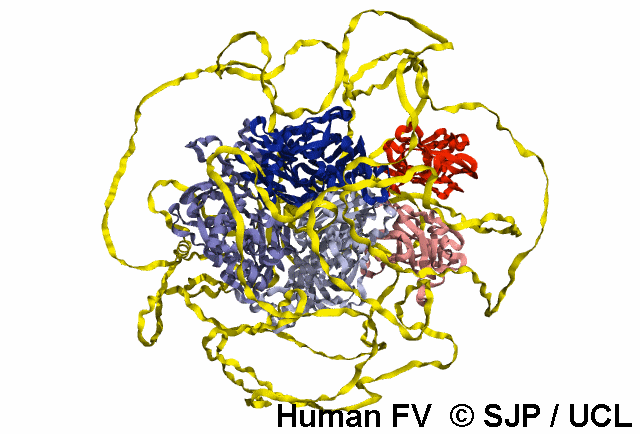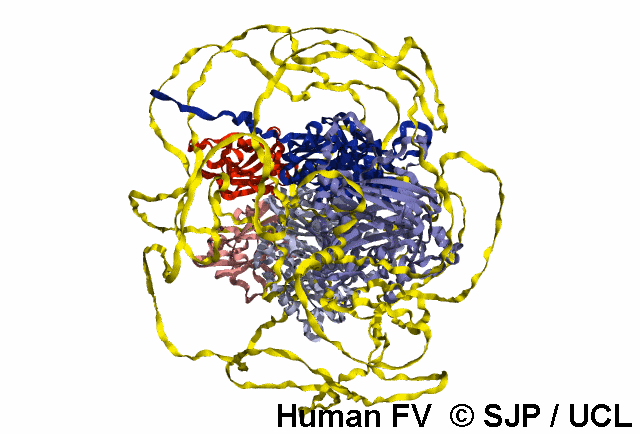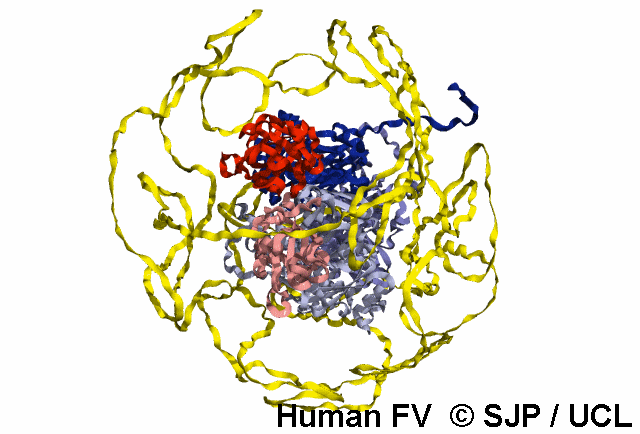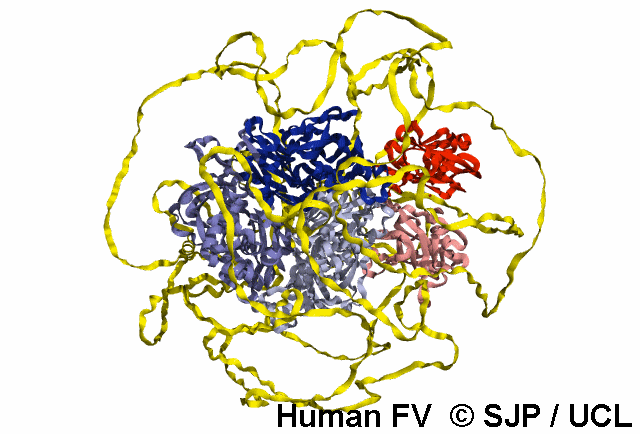

FV is a plasma glycoprotein that plays a key role in the coagulation cascade. Specifically, activated FV (FVa) is essential in the prothrombinase complex and thrombin amplification. Said complex cleaves prothrombin into thrombin, aiding formation of the platelet plug, critical in coagulation.
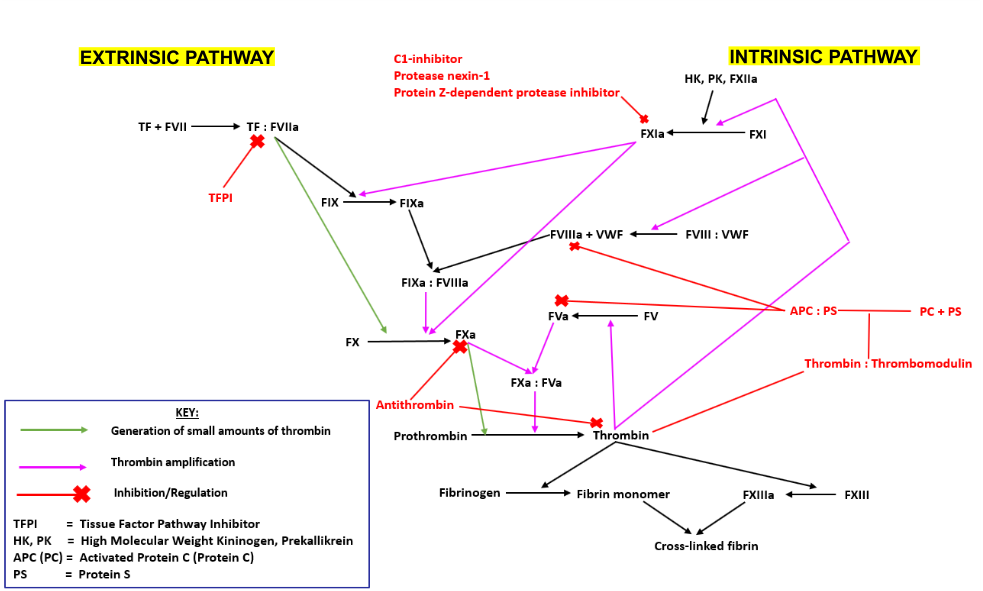
The coagulation cascade can be partitioned into the extrinsic and intrinsic pathways, driving thrombin production:
- The extrinsic pathway is initiated following vessel trauma, which results in the exposure of sub-endothelial tissue factor (TF) to the blood. TF forms a complex with activated Factor VII (FVIIa), which activates Factors IX (FIX) and X (FX). Activated FX (FXa) converts prothrombin into thrombin with cofactor FVa.
- The intrinsic pathway is initiated upon contact of endothelial collagen with Factor VII (FVII), which itself is complexed with Prekallikrein (PK) and High Molecular Weight Kininogen (HK). Activated FVII catalyses the activation of FVI, which then interacts with the extrinsic pathway, activating FIX. FIXa complexes with activated FVIII (FVIIIa) forming the intrinsic-tenase complex, which subsequently cleaves and activates FX.
- Another common pathway involves the thrombin-mediated conversion of fibrinogen into fibrin, required for stable clot formation. Thrombin also activates Factor VIII (FVIII) which cross-links fibrin, enhancing its resistance to dispersion by high blood pressure.
- In all cases, thrombin or FXa activate FV through proteolytic cleavage to FVa, resulting in a positive feedback loop whereby FVa and FXa generate additional thrombin.
FV is a single-chain glycoprotein consisting of domains in the order A1-A2-B-A3-C1-C2. FV shares sequence and structural homology with FVIII.
Note: The entirety of FV has yet to be crystallised, likely owing to its flexibility and unstructured nature, as well as the heavily glycosylated B domain.
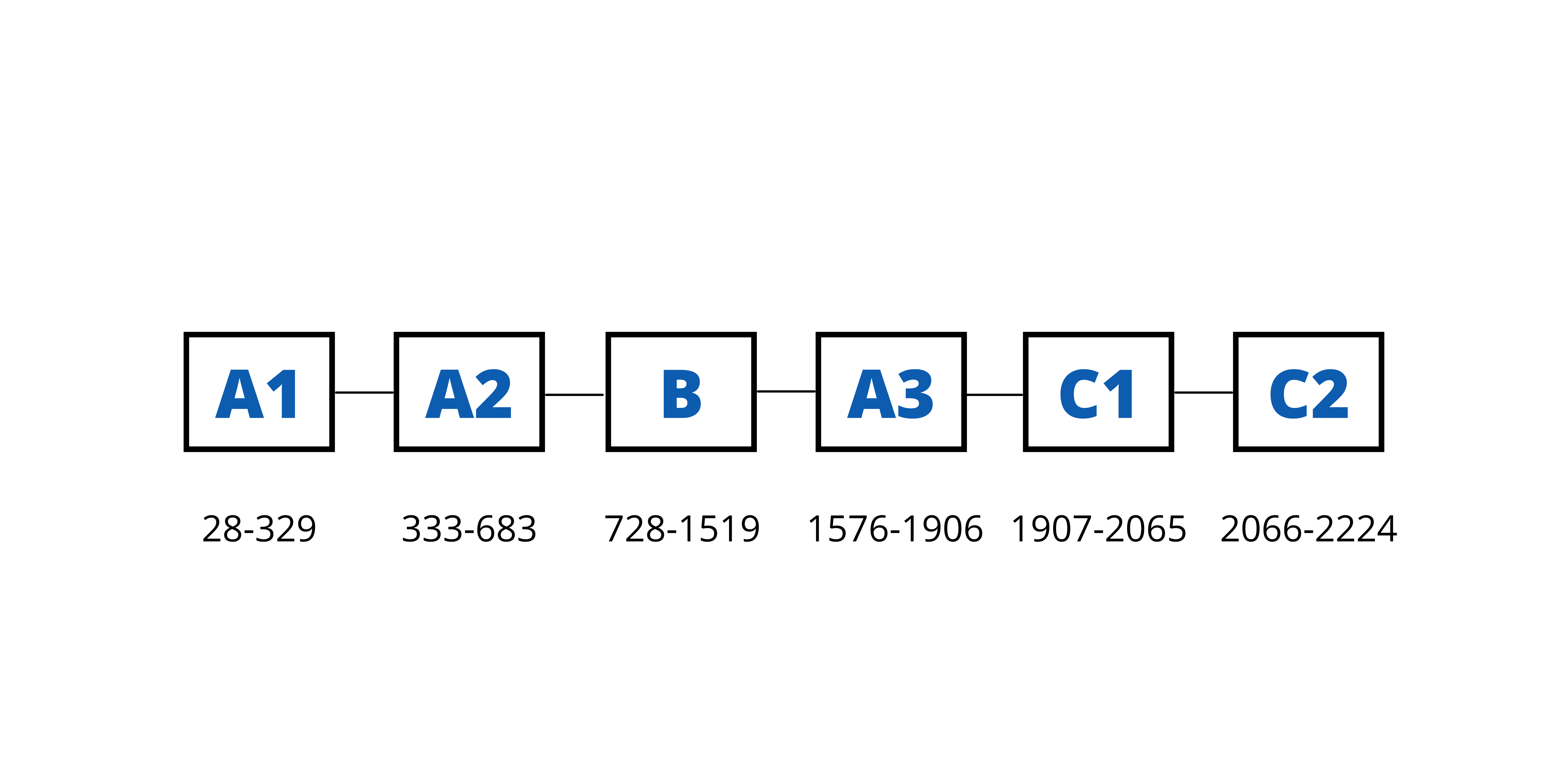
FV is synthesized in the liver and released into the blood in zymogenic form. Prior to release, the signal peptide is post-translationally modified and subsequently cleaved. FV circulates in the blood as a 6 domain protein with MW of 330 kDa. It can be divided into a heavy chain, consisting of A1 and A2, and a light chain, consisting of A3, C1, and C2.
FV can be activated either by FXa or thrombin. Both processes involve proteolytic cleavage of FV at R709, R1018, and R1545, releasing the B domain from the middle of the protein. The heavy and light chains remain associated through a single calcium ion. Cleavage results in a conformational change, enabling FVa to act as a cofactor for FXa to cleave prothrombin.
FXa alone has minimal affinity for the substrate prothrombin. However, once activated, FXa complexes with cofactor FVa, forming the prothrombinase complex, a complex with high prothrombin affinity. Prothrombin binds an exosite on the protease complex and is cleaved at two distinct sites, Arg320 and Arg271. Thrombin is subsequently released and facilitates platelet plug formation, preventing excess blood loss.
Variants within the F5 gene can cause several diseases including FV deficiency, FV Leiden thrombophilia, and F5 Atlanta.
FV coagulant activity (FV:C) in normal human plasma ranges from 60-140 IU/dL. Severely FV deficient patients have FV:C levels of < 1 % of normal, moderately deficient individuals have FV:C levels of 1-5 % of normal and mildly deficient individuals possess FV:C levels of 6-10 % of normal.
Individuals with severe FV deficiency are typically homozygous or compound heterozygous for causative mutations, whereas individuals who are partially deficient are heterozygous, with one mutated allele.
FV Leiden thrombophilia is associated with activated protein C (APC) resistance, whereby mutations in FV render it less susceptible to the inactivating effects of APC. This results in higher than normal coagulation due to increased thrombin production.
F5 Atlanta is characterized by a large deletion in exon 13, resulting in truncation of the FV protein.
Individuals with FV deficiency experience variable bleeding symptoms, the severity correlating with the level of protein activity. Individuals with lower FV:C will experience more severe symptoms than those with higher protein activity. Symptom onset can occur at any time, though severely affected individuals suffer from an earlier age.
- Mildly affected individuals experience minor symptoms such as easy bruising, nosebleeds, gum bleeding and hematuria (blood in urine). Such individuals are also prone to prolonged and excessive post-operative bleeding.
- Severely affected individuals suffer from more severe bleeds including hemarthrosis (bleeding into joints), intramuscular bleeds and hematomas. Additionally, such individuals are more susceptible to melena (blood in stool), bleeding in the gastrointestinal tract and intracranial hemorrhages.
- Females suffering from FV deficiency often experience menorrhagia (heavy or prolonged bleeding during menstruation). Such individuals are also at greater risk of pregnancy complications including miscarriage and post-partum hemorrhage (bleeding after pregnancy).
Those with FV Leiden often do not experience symptoms. However, the disease can cause abnormal blood clots which are associated with symptoms such as pain, swelling, rapid heartbeat, or shortness of breath. Symptoms depend on the location of the blood clot.
F5 Atlanta is very rare and therefore less well characterized, but severe bleeding has been observed in known patients.
Current available therapy for FV deficiency and F5 Atlanta consists of FV replacement and the use of antifibrinolytics/ fibrin glue.
FV Replacement: Coagadex, Prothrombin Complex Concentrates (PCC) and Fresh Frozen Plasma (FFP)
- Prothrombin Complex Concentrate (PCC) and Fresh Frozen Plasma (FFP) – PCC and FFP are concentrates containing FV along with other coagulation factors such as Factors II, VII and IX. FFP is the fluid portion of whole blood and also contains additional plasma proteins. PCC and FFP are administered intravenously into deficient individuals to replace FV and promote coagulation.
Note: Given that PCC and FFP are not FV specific, problems such as thrombosis can arise due to accumulation of coagulation factors that the individual is not deficient in. In addition, whilst proving effective, the use of FFP and PCC is often contra-indicated and is thus precluded in significant numbers of individuals. A FV concentrate, similar to Coagadex used to treat FX deficiency, is currently being tested.
Antifibrinolytics/ Fibrin Glue: Used to treat individuals with minor forms of FV deficiency.
- Antifibrinolytics - Antifibrinolytics are drugs that work to prevent clot breakdown within the body, minimising the risk of excess bleeding. Examples of antifibrinolytics include tranexamic acid and aminocaproic acid.
- Fibrin Glue - Fibrin is a protein involved in cross-linking and stabilising clots within the body. Fibrin glue is used externally to facilitate clot formation and stabilisation. Fibrin glue is most commonly used post-operatively in FV deficient individuals to facilitate wound healing and minimise the risk of prolonged post-operative bleeding.
Current available therapy for FV Leiden consists of blood thinning medications to reduce the risk of abnormal blood clots.
Heparin